
Increasingly, routine surveillance and monitoring of foodborne pathogens using whole-genome sequencing is creating opportunities to study foodborne illness epidemiology beyond routine outbreak investigations and case–control studies.

Using a global phylogeny of Salmonella entericaserotype Typhimurium, we found that major livestock sources of the pathogen in the United States can be predicted through whole-genome sequencing data. Relatively steady rates of sequence divergence in livestock lineages enabled the inference of their recent origins. Elevated accumulation of lineage-specific pseudogenes after divergence from generalist populations and possible metabolic acclimation in a representative swine isolate indicates possible emergence of host adaptation.
We developed and retrospectively applied a machine learning Random Forest classifier for genomic source prediction of Salmonella Typhimurium that correctly attributed 7 of 8 major zoonotic outbreaks in the United States during 1998–2013. We further identified 50 key genetic features that were sufficient for robust livestock source prediction.
Zoonotic source attribution of Salmonella Enterica serotype typhimurium using genomic surveillance data, United States
January 2019
Emerging Infectious Diseases vol. 25 no. 1
Shaokang Zhang, Shaoting Li, Weidong Gu, Henk den Bakker, Dave Boxrud, Angie Taylor, Chandler Roe, Elizabeth Driebe, David M. Engelthaler, Marc Allard, Eric Brown, Patrick McDermott, Shaohua Zhao, Beau B. Bruce, Eija Trees, Patricia I. Fields, and Xiangyu Deng
https://wwwnc.cdc.gov/eid/article/25/1/18-0835_article
 2008 (
2008 (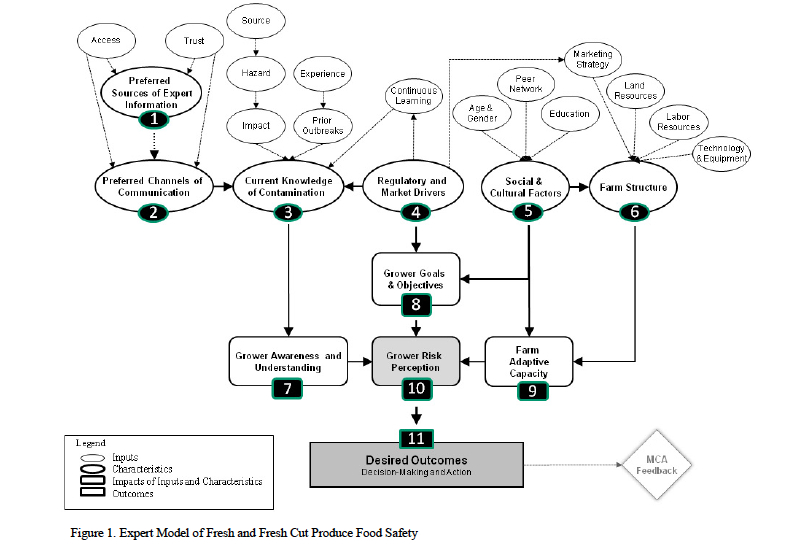 farm scale, marketing practices, and the need for appropriate food safety interventions. We further discuss how this loosely-coupled systems perspective can both aid our understanding of grower-decision-making and provide a basis for developing equitable solutions to on-farm food safety issues as part of a social-psychological approach to addressing these issues.
farm scale, marketing practices, and the need for appropriate food safety interventions. We further discuss how this loosely-coupled systems perspective can both aid our understanding of grower-decision-making and provide a basis for developing equitable solutions to on-farm food safety issues as part of a social-psychological approach to addressing these issues. Beef Cattle Production Systems and included four interrelated studies:
Beef Cattle Production Systems and included four interrelated studies: